
※BNT162b2のファクトシート(一部)
2021年2月14日に、ファイザーとバイオンテックが開発したCOVID-19用ワクチン候補「BNT162b2」が厚労省によって特例承認されました。 でも医薬品開発に関わった経験がある人でないと特例承認という言葉の意味がよくわからないと思います。 そこで補足として、医薬品の特例承認について少し説明したいと思います。
特例承認は医薬品医療機器等法第14条の3に規定されていて、次の条件を満足する医薬品候補について厚生労働大臣が特別に承認を認める制度です。
直近では2020年5月に、特例承認としてレムデシビルの対象疾病にCOVID-19を追加しました。 この時は、アメリカ食品医薬品局FDA(Food and Drug Administration)がCOVID-19に対するレムデシビルの緊急使用を認可したので、「日本と同等水準と認められる承認制度を有する欧米」として認められていたイギリス、フランス、ドイツ、カナダに、急遽、アメリカを追加して(政令162号)特例承認しました。
ファイザーのCOVID-19用ワクチン候補「BNT162b2」は、2020年12月にFDAによって緊急使用認可を受けました。 緊急使用認可EUA(Emergency Use Authorization)とは新薬申請NDA(New Drug Application)に基づく正式な承認ではなく、保健福祉省HHS(Department of Health and Human Services)が国民の保健衛生上の緊急事態下にあることを認定し、医薬品等の緊急使用を宣言することによって発行されます。 そして緊急使用認可には次のような規定があります。
以上、シチ面倒くさそうなことが色々と規定されていますが、要するに緊急使用認可を受けた医薬品は正式な商品ではなく、まだ開発中の医薬品候補であり、緊急時だけの特別試用なので、医療従事者も患者もそのことを十分に理解した上で慎重に使用する必要があるということです。
そして保健福祉省宣言が解除されると、緊急使用認可を受けた医薬品候補は未承認扱いになります。 そのため開発した製薬企業は緊急使用認可中も臨床試験を続行し、あらためて正式な承認を受ける必要があります。 事実、ファイザーは現在もBNT162b2の臨床試験を続行中です。 BNT162b2が1年足らずで使用できるようになった大きな要因は、通常の医薬品開発手順の途中で緊急使用認可を受けたからです。
BNT162b2がアメリカで緊急使用許可を受けたので、日本でも厚労省によって特例承認されました。 特例承認はアメリカの緊急使用認可と同様の緊急時の薬事対応であり、その概要は最初に書いたとおりです。 しかし特例承認は欧米からの外圧によって制定されたところがあるので、次のように色々と問題があります。

以上のことから、特例承認が欧米に甘くて日本国内に厳しい典型的な外圧による制度であることがわかると思います。 そしてBNT162b2については、アメリカの緊急使用認可が取り消されれば特例承認も取り消されることになると思います。
BNT162b2は、本家本元のアメリカでは仮名称ファイザー・バイオンテックCOVID-19ワクチン(Pfizer-BioNTech COVID-19 Vaccine)で流通しているのに対して、日本では正式な販売名コミナティ筋注(Comirnaty intramuscular injection)で流通します。 そして医療従事者は、このワクチンを使用する前に接種者または代諾者に最新の有効性と安全性について文章で説明した上で、予診票等で文書による同意を得る必要があります。 しかし接種者または代諾者にファクトシート等の詳細な資料を提供する義務はなく、しかも添付文書には特例承認の詳しい説明がありません。
これでは「特例承認されたワクチンは正式な商品ではなく、まだ開発中の医薬品候補であり、緊急時だけの特別試用なので、医療従事者も接種者もそのことを十分に理解した上で慎重に接種する必要がある」ということが周知徹底されない可能性が高いと思います。
アメリカの緊急使用認可では、当該医薬品が正式承認を受けるためには、緊急使用認可下で実施していた臨床試験データを用いてあらためて新薬申請を行う必要がある、と規定してあります。 しかし特例承認ではそういった規定が明確になっていません。 そのためアメリカでファイザー・バイオンテックCOVID-19ワクチン(コミナティ筋注)の緊急使用認可が取り消されれば日本の特例承認も取り消され、アメリカで正式承認になれば日本でもなし崩し的に正式承認になる可能性が高いと思います。
また特例承認申請用にファイザーが日本で実施した臨床試験は、BNT162b2接種群119例、プラセボ接種群41例の無作為化比較対照試験です。 その結果、SARSコロナウイルス2に対する抗体の発現状況と有害事象の発現状況がアメリカで実施された4万例の臨床試験の結果と矛盾しないので、「日本でも有効性と安全性が確認された」として特別承認を受けました。
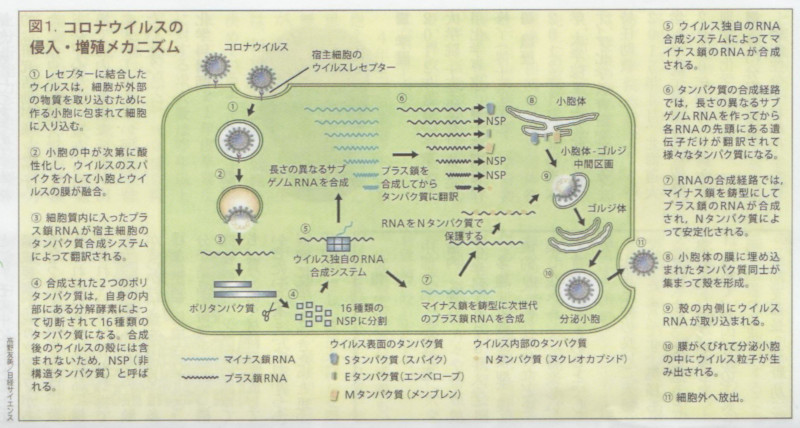
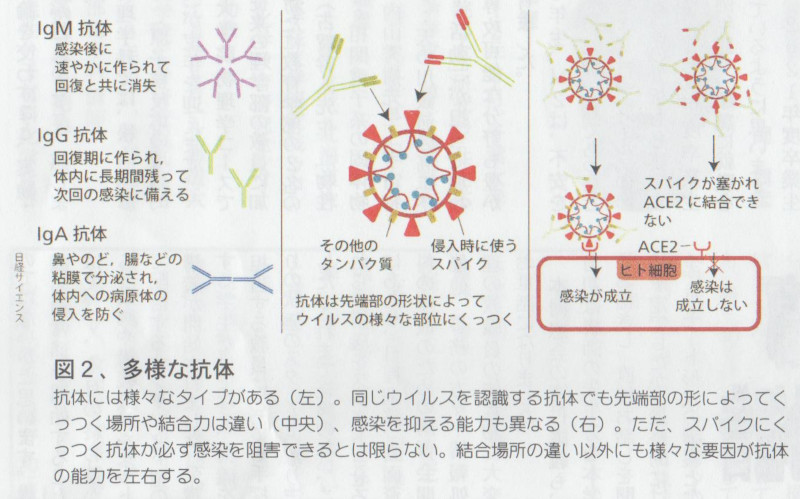
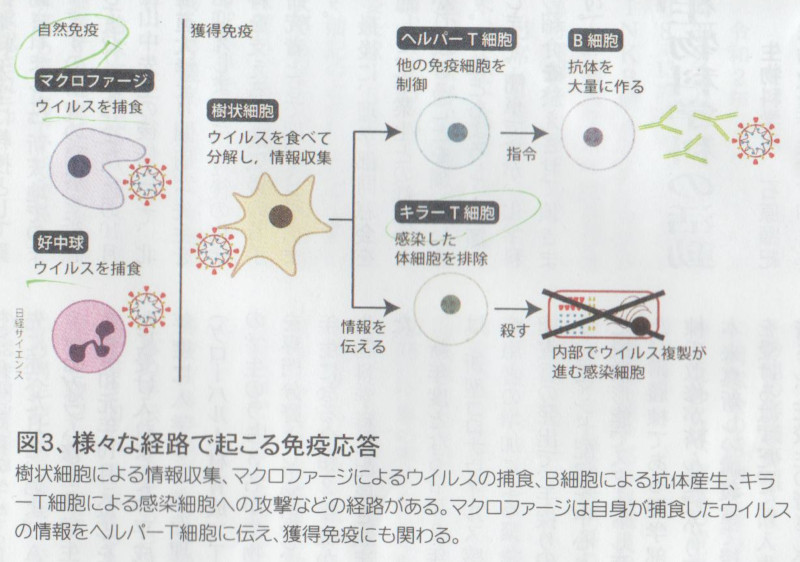
しかし抗体は感染予防を担う免疫システムの構成要素のひとつにすぎません。 免疫システムはウイルスを捕食するマクロファージや好中球、ウイルスを分解してその情報を収集する樹状細胞、樹状細胞から伝えられた情報に基づいて感染した体細胞を排除するキラーT細胞、同じく樹状細胞から伝えられた情報に基づいて抗体を産出するヘルパーT細胞とB細胞、そしてウイルスの表面にあるスパイクと結合し、ウイルスが細胞内に侵入するのを防ぐ様々な抗体(IgM、IgG、IgA等)といった、数多くの構成要素がお互いに連携して作動する、複雑かつ精緻なシステムです。
そのため抗体ができていなければ免疫システムがうまく作動せず、感染を予防するのが難しいことは確かですが、たとえ抗体ができていても、免疫システム全体がうまく構成されていなければ――例えばウイルスのスパイクとうまく結合できない抗体だったりしたら――やはり感染を予防するのは難しいのです。
つまり抗体ができることはCOVID-19予防効果の必要条件ではあっても、十分条件ではないのです。 これがCOVID-19予防効果の必要十分条件なら、わざわざ4万例もの臨床試験を実施する必要はありません。 そしてアメリカで4万例もの臨床試験を実施したのに対して、アメリカの40分の1程度の感染率である日本において、わずか160例の臨床試験で「有効性と安全性が確認された」と考える医学研究者がいたとしたら、僕はその人の研究のお手伝いをするのはお断りします。
ファイザーが日本で実施した160例の臨床試験は、とりあえず安全性を検証するための必要最小限のものであり、多分に政治的な判断がらみで実施されたものだと思います。 パンデミックという緊急事態下であることを考えると、これはある程度は致し方ないことだと思います。 でも今後のことを考えると、やはり有効性と安全性を厳密に検証するための大規模な臨床試験を実施する必要があると思います。 しかし残念ながら、現在の特例承認にはそのような臨床試験が必要であるとは規定されていません。 データ解析屋としては、この点が現在の特例承認制度の最大の問題点だと思います。
最後に「日本医師会 COVID-19 有識者会議声明 2020年5月17日」の「新型コロナウィルス感染パンデミック時における治療薬開発についての緊急提言」中に書かれている、次の言葉をあらためて紹介しておきます。
「しかし有事だからエビデンスが不十分でも良い、ということには断じてならない。
…
そして『科学』を軽視した判断は最終的に国民の健康にとって害悪となり、汚点として医学史に刻まれることになる。」


 webmaster@snap-tck.com
webmaster@snap-tck.com